Summary:
The equilibrium state of a reacting system is achieved
after a sufficiently long time, which depends on the rate of reaction. If the reaction is
irreversible (additive, decomposition,etc.) the equilibrium composition of the reacting
mixture is uniquely defined by stoichiometry because all reactants disappear at
equilibrium (see chapter 4). Reversible chemical equations R+S<=>P+Q
are
characterised by an equilibrium composition, expressed by equilibrium constants
K c =[P][Q]/([R][S]) or
K p =p P p Q /p R p
S
in terms of concentrations or partial pressures
respectively. Given K c the composition of the mixture can be calculated from
stoichiometry: For example, five unknown equilibrium concentrations [R],[S],[P],[Q]
and the reaction progress [z] corresponding to the balanced chemical equation
R+S<=>P+Q are determined by five equations [R]=[R] 0 -[z], [S]=[S] 0 -[z], [P]=[P]
0 +[z],
[Q]=[Q] 0 +[z], and Kc=[P][Q]/([R][S]). Substituting [R], [S], [P], [Q] into Kc a quadratic
equation for the reaction progress [z] is obtained and can be solved. Reversibly,
given the concentration of any species at equilibrium, the equilibrium constant Kc can
be calculated: Given for example the concentration [R] at equilibrium, the reaction
progress is [z]=[R] 0 -[R]. Then all the remaining concentrations [S],[P],[Q], as well as
the equilibrium constant Kc, are calculated by the substitution of [z] into their
definitions (this is an experimental method of Kc evaluation). Equilibrium constant Kp
can also be calculated theoretically, from the standard Gibbs energy change as
K p =exp(-G 0 /RT) and K c can be recalculated from K p by using an equation of state.
The rate of a chemical reaction is defined for any species participating in the
reaction as the amount of reacted species (in moles) within a unit time and unit
volume (d[R]/dt); given d[R]/dt the reaction rates of other species S,P,Q can be
calculated from the reaction stoichiometry. The basic equation used for estimating
the reaction rate is d[R]/dt=-k[R]
for monomolecular reactions, R->P+Q, while the
differential equation d[R]/dt=-k[R][S]
describes bimolecular reactions, R+S->P+Q.
Note that the products (species on the right hand side) have no effect on the reaction
rate. If the chemical equation is neither mono- nor bimolecular (which is difficult to
find at first glance, and only experiment can verify a guess) the summary reaction
should be decomposed to elementary reactions and the reaction course computed
by solving a system of differential equations, which describe the individual
elementary reactions. The solution can be simplified by careful examination of the
elementary reactions - the fastest of them can be substituted by the equilibrium
equations, thus reducing the number of differential equations.
If the components S,T,... involved in reaction R+S+T+...->... are in sufficient
excess, their concentrations remain approximately constant during reaction and we
can assume that the rate equation depends on the concentration of R only: d[R]/dt=-
k[R]n,
where n is the reaction order and k is the rate coefficient. The rate coefficient is
not a constant, k depends on temperature T, and the simplest form of this
relationship is the Arrhenius equation k=A×exp(-Ea/RT),
where A is frequency factor,
Ea activation energy of reaction, R is the universal gas constant and T is the
thermodynamic temperature. More advanced theories (e.g., collision theory) and
experiments lead to a slightly different expression for temperature dependence,
usually in the form k=ATb×exp(-Ea/RT), where exponent b is tabulated (see
appendix). Probably the best source of kinetic data are computerised databases,
available on CD-ROMs (e.g. NIST /National Institute of Standards and Technology/
Chemical Kinetics Database, see Internet address: srdata@nist.gov).
Activation energy Ea is a barrier which must be overcome so that the colliding
molecules can react. The activation energy Ea depends on the bonding energy of the
so called activated complex, a temporary molecule having only partially formed
bonds. The smaller the energy of the activated complex the higher the probability
that a collision will result in a chemical change. There are usually many ways to
decompose a summary chemical reaction into intermediate elementary reactions
(e.g., decomposition of reactants to free elements - radicals and subsequent forma-
tion of products). Every elementary reaction has its own activation energy and the
actual reaction mechanism (sequences of elementary reactions) leads through the
valley of the lowest activation energies. Sometimes species not explicitly enumerated
in lists either of reactants or products take part in the intermediate reactions. These
species, which are not consumed in the overall chemical reaction, are called
catalysts. Catalysts open other possible reaction paths, characterised by lower
activation energies, and thus their presence increases the overall reaction rate.
Rate equations describing the rates of forward and reverse reactions can also
be used for determining the steady state (i.e., for calculating equilibrium constants).
Comparing Kp, expressed by the rate equations, with the expression based on
thermodynamic analysis of equilibrium Kp=exp(-DG/RT), we come to the conclusion
that: DG-> =Ea-> -Ea (Gibbs energy change corresponding to the forward reaction
equals the difference of activation energies of forward and reverse reactions) and
A-> =A, the frequency factor of the forward reaction equals the frequency factor of
the reverse reaction.
The chemical reaction equilibrium and the rate of chemical reaction are
influenced by temperature and pressure. While a temperature increase always
speeds up reactions, it shifts the reversible reaction equilibrium to the right only if the
reaction is endothermic (this is an example of the Le Chatelier principle). Catalysts
increase reaction rates, too, but they have no effect on the equilibrium.
Knowledge of chemical kinetics is the basis of reaction engineering. We want
to know the composition of a reaction mixture in a batch reactor at a particular time.
The answer is a solution of the rate equation, giving a time dependence of the
reaction progress z(t). This result can also be used for the yield prediction of a
continuous reactor, if the time that the material spends in the reactor (residence
time) is known. Because the residence time is only exceptionally uniform for all the
particles flowing through the reactor, the mean reaction progress at the reactor outlet
must be calculated by integration across all possible residence times (usually from 0
/some particles find the way out very quickly, a so called short-cut/ to infinity /there
are also some unhappy particles helplessly wandering in the reactor or particles
trapped in a "Maelstrom swirl", in the so called dead regions/), so that
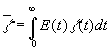
, where z(t) is the reaction progress, and E(t)
is the Residence Time Distribution depending on the flow structure inside the reactor.
The most frequently used functions E(t) are given in the following table, which is not
intended for learning, rather for calculation of examples:

All these models have only one parameter (N or Pe /the so called Peclet number/)
which characterises a particular reactor and which can be determined, e.g.,
experimentally. Some other details and models can be found in
Žitný, Thýn (1996).
·
@TEC: 3. 3.2003
Change language to


 peoples
peoples


 mailto: Zitny
mailto: Zitny